Blue Line Walkthrough - DNA Barcoding
DNA barcodes allow non-experts to objectively identify species – even from small, damaged, or industrially processed material. Just as the unique pattern of bars in a universal product code (UPC) identifies each consumer product, a "DNA barcode" is a unique pattern of DNA sequence that identifies each living thing. Short DNA barcodes, about 700 nucleotides in length, can be quickly processed from thousands of specimens and unambiguously analyzed by computer programs.
You can analyze relationships between DNA sequences by comparing them to a set of sequences you have compiled yourself, or by comparing your sequences to other that have been published in databases such as GenBank (National Center for Biotechnology Information). Generating a phylogenetic tree from DNA sequences derived from related species can also allow you to draw inferences about how these species may be related. By sequencing variable sections of DNA (barcode regions) you can also use the Blue Line to help you identify an unknown species, or publish a DNA barcode for a species you have identified, but which is not represented in published databases like GenBank http://ww.ncbi.nlm.nig.gov/genbank.
Wet-Lab Protocol
Though not covered in this tutorial, the wet-lab protocol and many additional resources are available:
Create Project
- Log-in to DNA Subway dnasubway.iplantcollaborative.org
- Click ‘Determine Sequence Relationships.’ (Blue Square)
- Select project type ‘Barcoding: rbcL.’
- Select sample sequence ‘rbcL sample 1.’
- Provide your project with a title, then Click ‘Continue.’ Alternatively, if you have sequenced your DNA using your Genewiz account, Select ‘Import trace files from DNALC.’ – Then select sequences to import.
View Sequence Data
- Click ‘Sequence Viewer’ to show a list of your sequences.
- Click on a sequence name to show the sequences’ trace file.
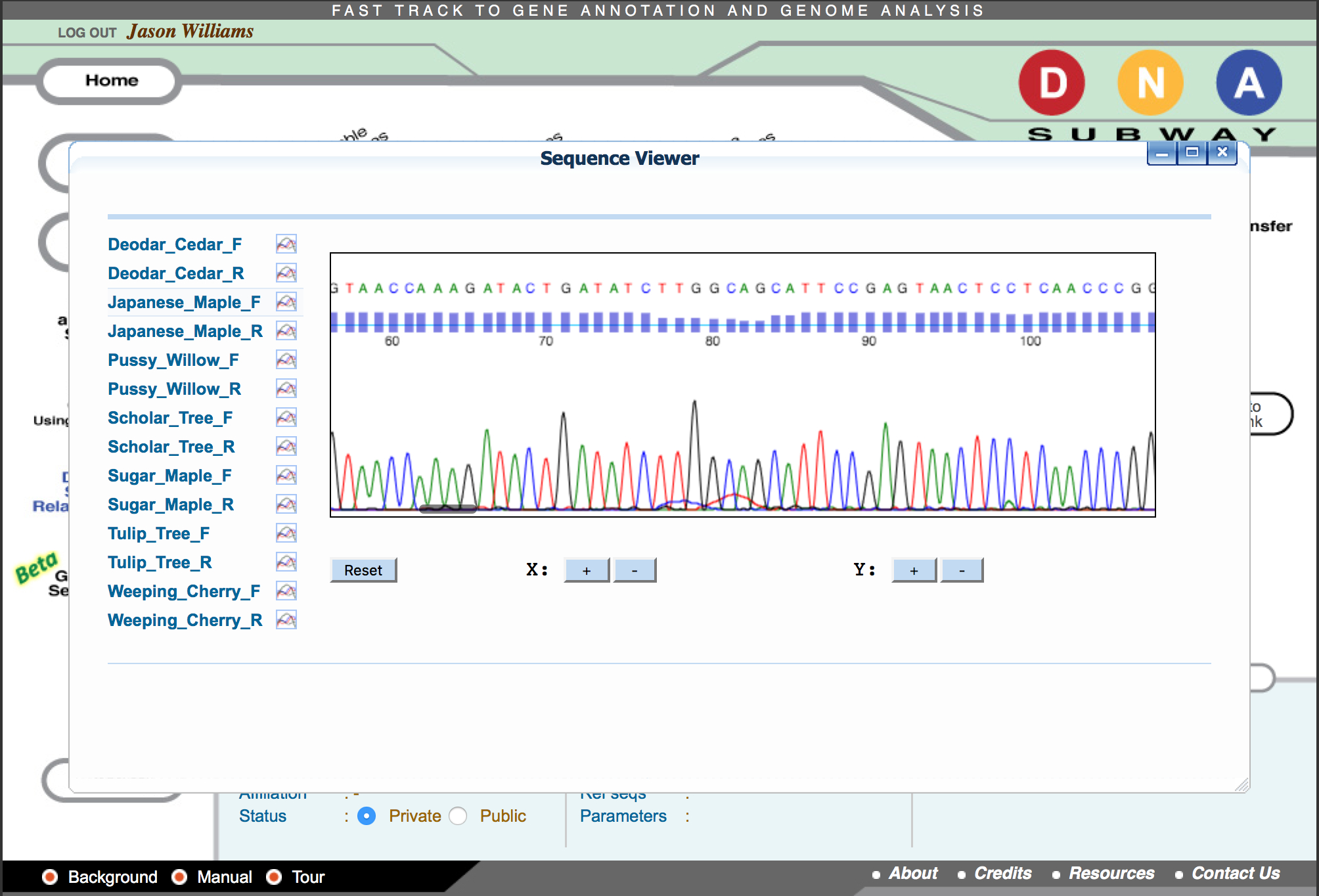
Questions
- What do you notice about the electropherogram peaks and quality scores at nucleotide positions labeled “N”?
- Where do the ‘N’s’ in the sequence tend to be distributed, and Why?
Trim sequence,reverse complement and pair
By default, DNA Subway assumes that all reads are in the forward orientation, and displays an ‘F’ to the right of the sequence. If any sequence is not in that orientation, click the “F” to reverse compliment the sequence. The sequence will display an “R” to indicate the change.
- Click ‘Sequence Trimmer.’
- Click ‘Sequence Trimmer’ again to examine to changes made in the sequence
- Click ‘Pair Builder.’
- Select the check boxes next to the sequences that represent bidirectional reads of the same sequence set. Alternatively Select the ‘Auto Pair’ function and verify the pairs generated.
- As necessary, Reverse Compliment sequences that were sequenced in the reverse orientation by clicking the ‘F’ next to the sequence name. The ‘F’ will become an ‘R’ to indicate the sequence has been reverse complimented.
- Save the created pairs.
Build a consensus sequence
This step remove poor quality areas at the 5’ and/or 3’ ends of the consensus sequence.
- Click on “Trim Consensus.” Scroll left and right in the consensus editor window to identify what string of nucleotides from the consensus sequence you want to trim.
- Click on the last consensus sequence nucleotide that you want to trim. A red line will indicate what nucleotides will be removed from the consensus sequences.
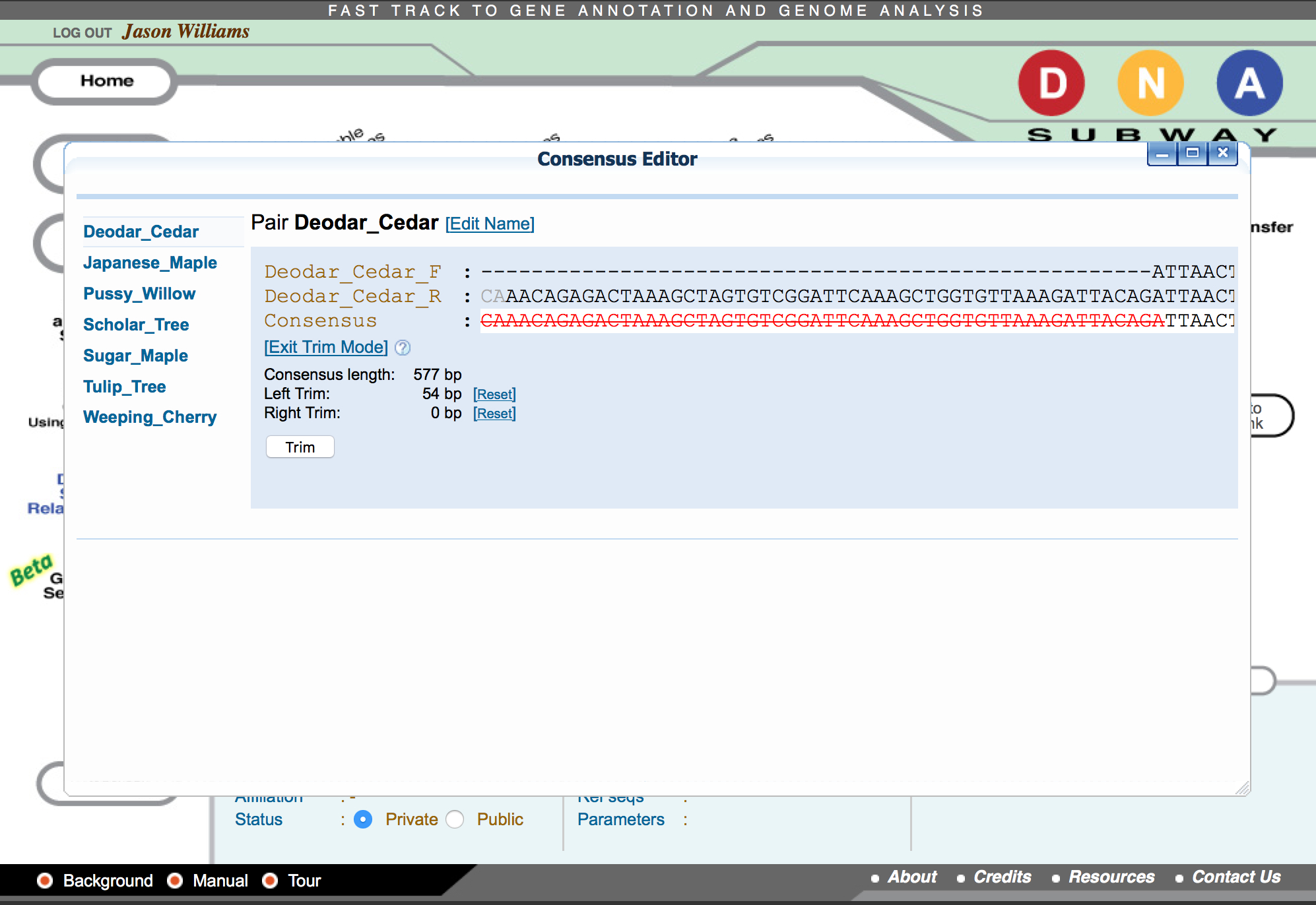
- Click “Trim.” A new “Consensus Editor” window will pop up displaying the trimmed sequences.
Check for matches in GenBank
- Click ‘BLASTN' then click the 'BLAST' link to BLAST the sequence of interest. When the search is completed a 'View' link will appear.
- Examine the BLAST matches for candidate identification. Clicking the species name given in the BLAST hit will also give additional information/photos of the listed species.
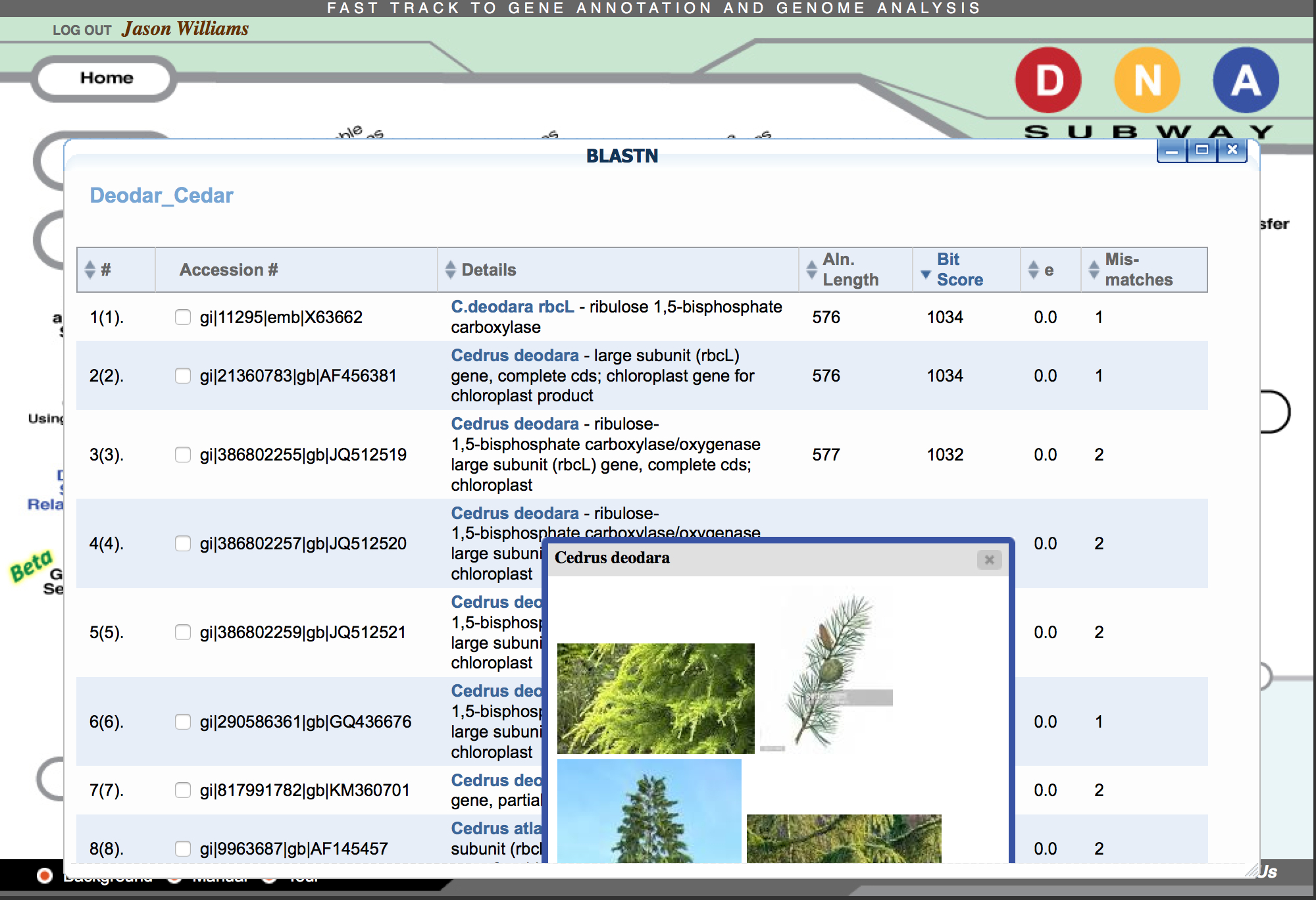
- If desired, select the check box next to any hit, and select ‘Add BLAST hits to project’ to add selected sequences to your project.
Questions
- BLAST will return the closest matches present in GenBank. Will you be able to identify an unknown species using BLAST alone? Why or Why not?
Add Reference data
Depending on the project type you have created, you will have access to additional sequence data that may be of interest. For example, if you are doing a DNA barcoding project using the rbcL gene, samples of rbcL sequence from major plant groups (Angiosperms, Gymnosperms, etc.) will be provided. Choose any data set to add it to your analysis; you will be able to include or exclude individual sequences within the set in the next step.
- Click ‘Reference Data.’
- Select 'Common plants'
- Click ‘Add ref data’ to add the data to your project.
Build a multiple sequence alignment and phylogenetic tree
Select Data
- Click ‘Select Data.’
- Select any and all sequences you wish to add to your tree.
- Click ‘Save.” to select data
- Click ‘MUSCLE.’
- Click ‘MUSCLE’ again to open the sequence alignment window.
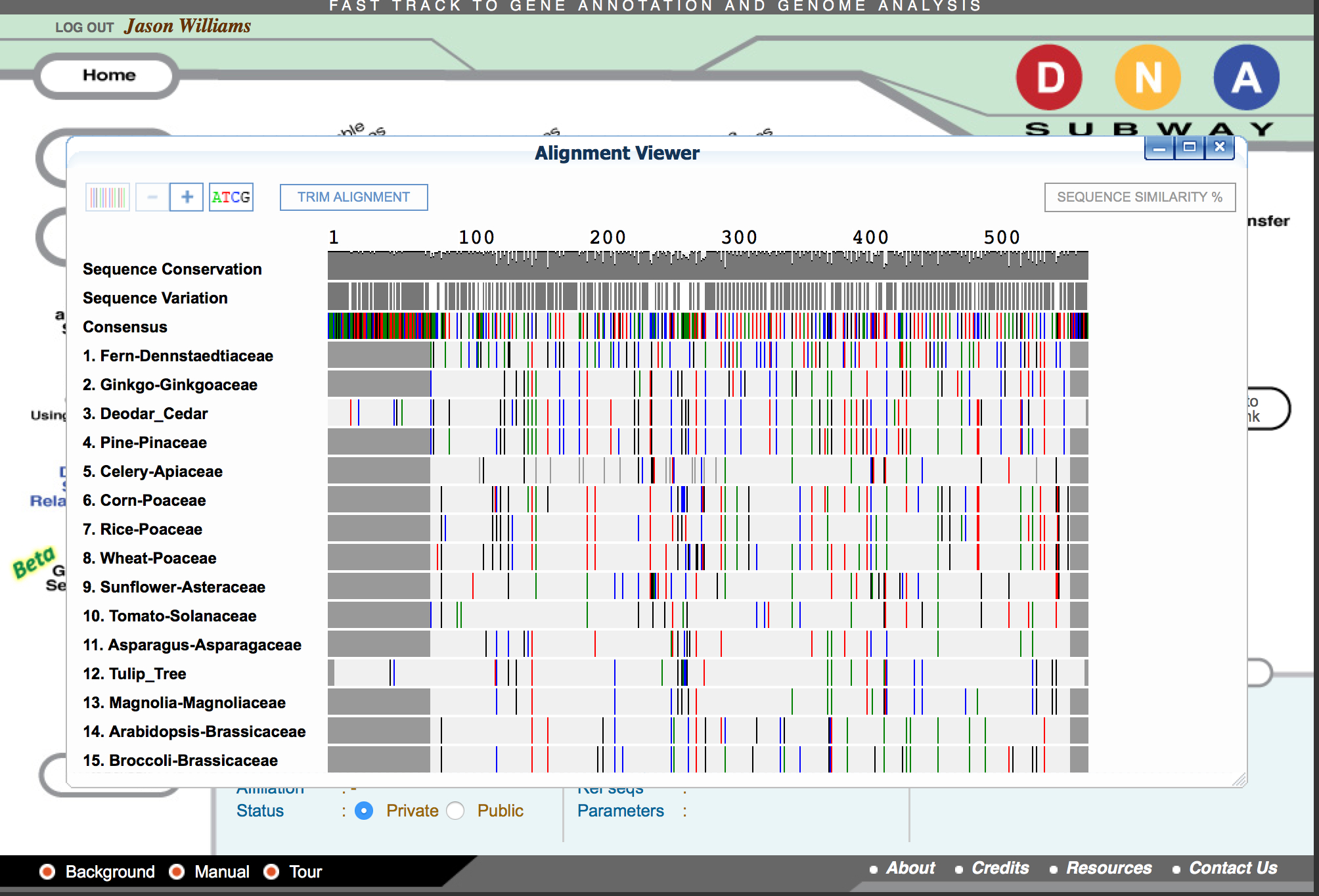
- Examine the alignment and then select the 'Trim Alignment' link in the upper-left of the Alignment viewer'
Build phylogenetic tree
- Click 'PHYLIP NJ' and then click again to examine a neighbor-joining tree
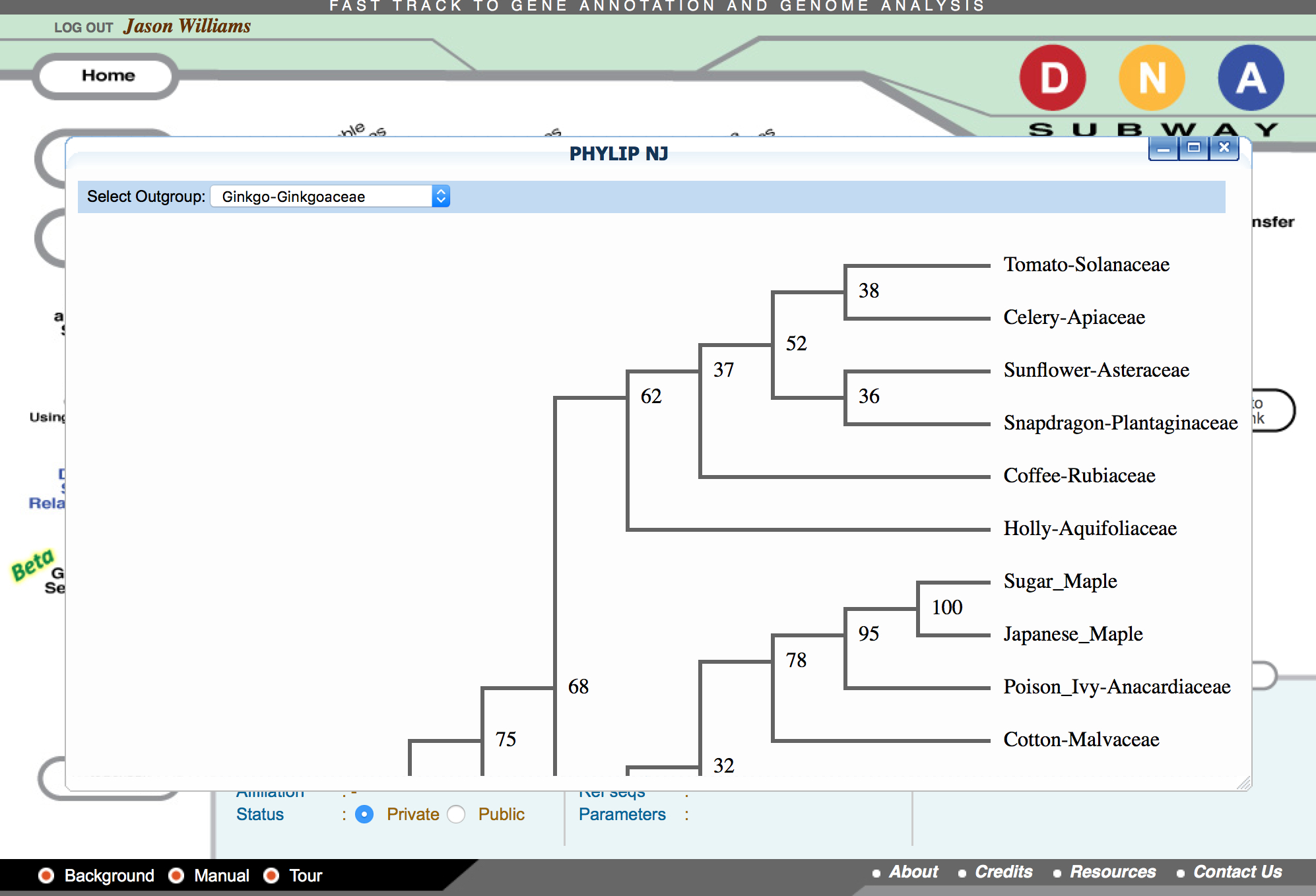
- Click 'PHYLIP ML' and then click again to examine a maximum-likelihood tree
Questions
- What relationship do you see between sequences that have more mutations (align less well with majority of sequences) in the alignment and the lengths of a sequences’ branch on the tree?
- Do you see differences in the phylogenetic tree generated by the Neighbor-joining vs. Maximum likelihood method?